
Wilson Disease: A Story of Copper and Time
How a silent genetic error slowly injures the liver—and why early recognition can change a life, and sometimes save one.
Where the Story Begins
Wilson disease does not announce itself loudly. It begins quietly—at birth—with a pair of altered genes that change how the liver handles copper. For years, sometimes decades, nothing seems wrong. Life moves forward. School, work, families. Meanwhile, copper accumulates, cell by cell, inside the liver [1].
The Hidden Gene
The culprit is ATP7B, a gene responsible for packaging excess copper into bile. In Wilson disease, that process fails. Copper is not excreted—it is stored. Parents are usually unaware carriers, healthy themselves, unknowingly passing along a condition that will surface much later [2].
The Liver Is Usually First
The liver bears the earliest and greatest burden. In some, this appears as mild lab abnormalities. In others, inflammation accelerates, leading to fibrosis, cirrhosis, portal hypertension, and complications familiar to every hepatologist [3].
In children and young adults, the story can turn abruptly—acute liver failure, hemolysis, confusion—an emergency chapter that demands immediate recognition [4].
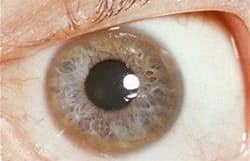
Clues Along the Way
Wilson disease is often misread. Tremor may be blamed on anxiety. Mood changes on stress. A Kayser–Fleischer ring—when looked for—can become the visual clue that reframes the entire narrative [5].
Diagnosis: The Turning Point
Diagnosis is rarely a single test. It is a convergence: low ceruloplasmin, elevated urine copper, genetic confirmation, and clinical judgment. When the diagnosis is made early, the trajectory changes—from decline to control [2].
A Lifelong Treatment Path
Chelation removes copper. Zinc blocks absorption. Treatment is not optional and not temporary. Stopping therapy allows copper to reclaim lost ground—and often does so quickly [3].
When Transplant Becomes the Next Chapter
For patients with acute liver failure or irreversible cirrhosis, liver transplantation becomes the defining chapter. Unlike many liver diseases, transplant in Wilson disease corrects the metabolic defect. The new liver knows how to handle copper [6].
The Ripple Through Families
One diagnosis often uncovers others. Siblings, children, relatives—screening becomes prevention. In Wilson disease, finding one patient can quietly save several more [2].
References
- EASL. Clinical Practice Guidelines: Wilson’s disease. J Hepatol. Link
- AASLD Practice Guidance: Wilson Disease. Link
- Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease. Hepatology. PubMed
- Camarata MA et al. Acute liver injury in Wilson disease. PubMed
- NIDDK. Wilson disease overview. Link
- Ferrarese A et al. Liver transplantation for Wilson disease. PubMed